
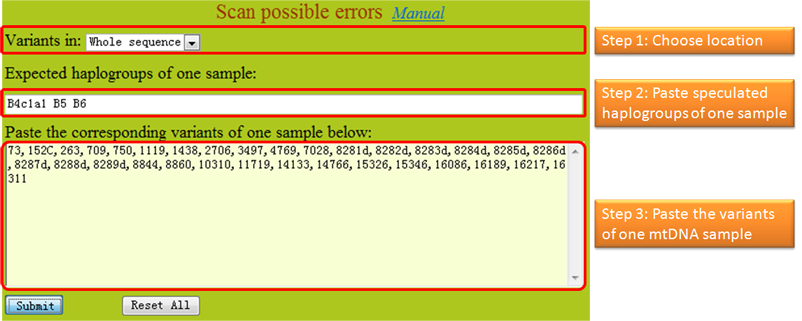

Tips:
1. All of the sequence variations identified by users should follow International Society for Forensic Genetics guidelines (reference). The sequence variations can have one of the four different styles as exemplified in the first step. The program checks one sample per run. 2. In MitoTool, the insertions are recorded with a style like "100+C" or "100insC"; the lone deletions are recorded with a style like "249d", "249del" or "249delA"; the continual deletions are recorded with a style like "290-291d" or "290-291del"; the transversions are recorded with a style like "16182C" or "A16182C"; the transitions are recorded with a style like "73" or "A73G"; each variant needs to be separated by a comma or a space. 3. The program can accept multiple haplogroup assignments of one mtDNA and export the potential missing variants for each claimed haplogroup, which needs to be separated by a comma, a space or a semicolon. Variants from one sample need to be inputted in one line without inserting a line break in the middle. 4. Due to phylotree updated, we recommend user inputted these following variants like the later style. 573+C(n) => 573+XC or 573insXC 960+C(n) => 960+XC or 960insXC 965+C(n) => 965+XC or 965insXC 5899+C(n) => 5899+XC or 5899insXC 8278+C(n) => 8278+XC or 8278insXC